gga_load_data tools
The gga_load_data tools allow automated deployment of GMOD visualisation tools (Chado, Tripal, JBrowse, Galaxy) for a bunch of genomes and datasets. They are based on the Galaxy Genome Annotation (GGA) project.
A stack of Docker services is deployed for each species, from an input yaml file describing the data.
See examples/example.yml for an example of what information can be described and the correct formatting of this input file.
The services currently deployed are:
- Chado database
- Tripal: database interface and hub to all applications
- Elasticsearch: searching service used in Tripal
- JBrowse: genome browser
- Nginx proxy: page to download the data files
- Blast (optional): BLAST interface to query the data
- Galaxy: data loading orchestrator for administrators
A GGA environment is deployed for each different species at https://hostname/sp/genus_species/. Multiple strains can have the same species and are deployed in the same GGA environment.
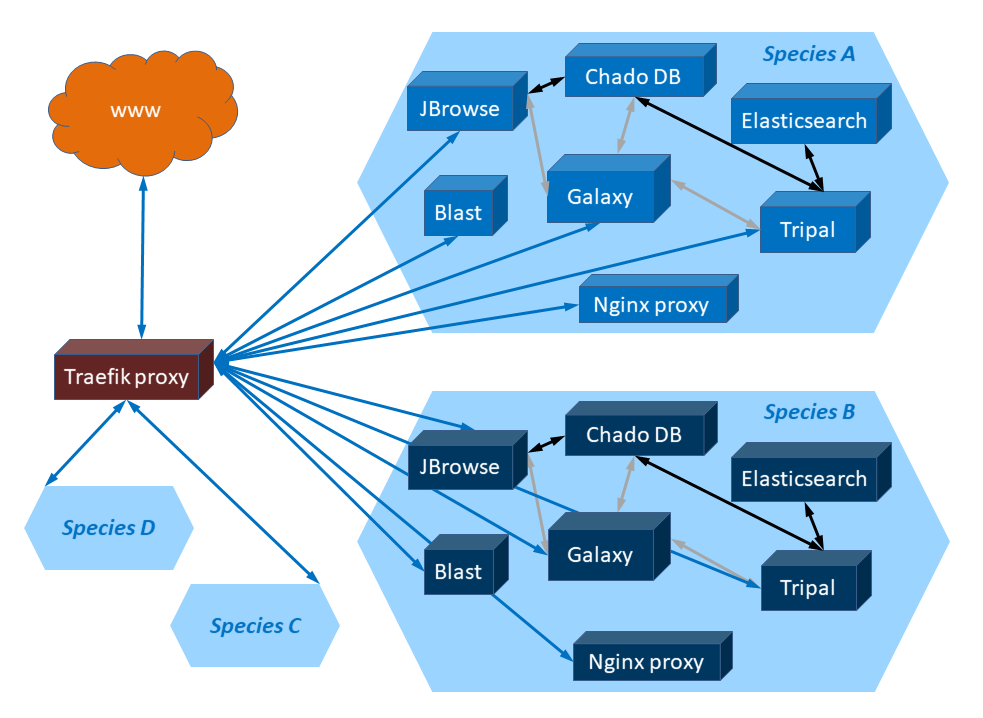 Figure : Schematic representation of a set of Docker containers deployed with
GGA for typical genomes. Cuboids represent Docker containers. Hexagons represent different
sets of Docker containers. Blue arrows represent HTTP traffic. Gray arrows represent
data exchange performed using Galaxy tools. Black arrows represent data exchange
inherent in applications.
Figure : Schematic representation of a set of Docker containers deployed with
GGA for typical genomes. Cuboids represent Docker containers. Hexagons represent different
sets of Docker containers. Blue arrows represent HTTP traffic. Gray arrows represent
data exchange performed using Galaxy tools. Black arrows represent data exchange
inherent in applications.
Requirements
To run the gga_load_data tools, Python 3.6 and the packages listed in requirements.txt are required.
To deploy the GGA Docker services, one or multiple hosts machines are required with Docker installed, and a swarm (for cluster management and orchestration).
Reverse proxy and authentication
Traefik is a reverse proxy which allows to direct HTTP traffic to various Docker Swarm services. The Traefik dashboard is deployed at https://hostname/traefik/
Authelia is an authentication agent, which can be plugged to an LDAP server, and that Traefik can used to check permissions to access services.
The authentication layer is optional. If used, the config file needs the variables https_port, authentication_domain_name, authelia_config_path, authelia_secrets_env_path, authelia_db_postgres_password.
Authelia is accessed automatically by Traefik to check permissions everytime someones wants to access a page. If the user is not logged in, he is redirected to the authelia portal. Note that Authelia needs a secured connexion (no self-signed certificate) between the upstream proxy and Traefik (and https between internet and the proxy).
Steps
The "gga_load_data" tools are composed of 4 scripts:
- gga_init: Create directory tree for organisms and deploy stacks for the input organisms as well as Traefik and optionally Authelia stacks
- gga_get_data: Create
src_datadirectory tree for organisms and copy datasets for the input organisms intosrc_data - gga_load_data: Load the datasets of the input organisms into their Galaxy library
- run_workflow_phaeoexplorer: Remotely run a custom workflow in Galaxy, proposed as an "example script" to take inspiration from as workflow parameters are specific to the Phaeoexplorer data
Usage:
For all scripts one input file is required, that describes the species and their associated data.
(see examples/citrus_sinensis.yml). Every dataset path in this file must be an absolute path.
Another yaml file is required, the config file, with configuration variables (Galaxy and Tripal passwords, etc..) that
the scripts need to create the different services and to access the Galaxy container. An example of this config file is available
in the examples folder.
The input file and config file have to be the same for all scripts!
- Deploy stacks part:
$ python3 /path/to/repo/gga_init.py input_file.yml -c/--config config_file.yml [-v/--verbose] [OPTIONS]
--main-directory $PATH (Path where to create/update stacks; default=current directory)
--force-traefik (If specified, will overwrite traefik and authelia files; default=False)- Copy source data file:
$ python3 /path/to/repo/gga_get_data.py input_file.yml [-v/--verbose] [OPTIONS]
--main-directory $PATH (Path where to access stacks; default=current directory)- Load data in Galaxy library and prepare Galaxy instance:
$ python3 /path/to/repo/gga_load_data.py input_file.yml -c/--config config_file.yml [-v/--verbose]
--main-directory $PATH (Path where to access stacks; default=current directory)- Run a workflow in galaxy:
$ python3 /path/to/repo/run_workflow_phaeoexplorer.py input_file.yml -c/--config config_file --workflow workflow_type [-v/--verbose] [OPTIONS]
--workflow (Valid options: "chado_load_fasta_gff_jbrowse", "blast", "interpro", preset workflows are available in the "workflows_phaeoexplorer" directory)
--main-directory $PATH (Path where to access stacks; default=current directory)The data loading into Galaxy with gga_load_data.py should be run only once the Galaxy service deployed with gga_init.py is ready.
The gga_load_data.py script checks that the Galaxy service is ready before loading the data and exit with a notification if it is not.
The status of the Galaxy service can be checked manually with $ docker service logs -f genus_species_galaxy or
./serexec genus_species_galaxy supervisorctl status.
Note:
When deploying the stack of services, the Galaxy service can take a long time to be ready, because of the data persistence.
In development mode only, this can be disabled by setting the variable galaxy_persist_data to False in the config file.
Directory tree:
For every input organism, a dedicated directory is created with gga_get_data.py. The script creates this directory and all subdirectories required.
If the user is adding new data to a species (for example adding another strain dataset to the same species), the directory tree will be updated
Directory tree structure:
/main_directory
|
|---/genus1_species1
| |
| |---/blast
| | |---/links.yml
| | |---/banks.yml
| |
| |---/nginx
| | |---/conf
| | |---/default.conf
| |
| |---/blast
| | |---/banks.yml
| | |---/links.yml
| |
| |---/docker_data # Data used internally by docker (do not delete!)
| |
| |---/src_data
| | |---/genome
| | | |---/genus1_species1_strain_sex
| | | |---/vX.X
| | | |---/vX.X.fasta
| | |
| | |---/annotation
| | | |---/genus1_species1_strain_sex
| | | |---/OGSX.X
| | | |---/OGSX.X.gff
| | | |---/OGSX.X_pep.fasta
| | | |---/OGSX.X_transcripts.fasta
| | |
| | |---/tracks
| | |---/genus1_species1_strain_sex
| |
| |---/apollo
| | |---/annotation_groups.tsv
| |
| |---/docker-compose.yml
| |
|---/traefik
|---/docker-compose.yml
|---/authelia
|---/users.yml
|---/configuration.yml