-
Loraine Guéguen authorede9a0a824

gga_load_data tools
The gga_load_data tools allow automated deployment of GMOD visualisation tools (Chado, Tripal, JBrowse, Galaxy) for a bunch of genomes and datasets. They are based on the Galaxy Genome Annotation (GGA) project.
A stack of Docker services is deployed for each species, from an input yaml file describing the data.
See examples/example.yml for an example of what information can be described and the correct formatting of this input file.
The services currently deployed are:
- Chado database
- Tripal: database interface and hub to all applications
- Elasticsearch: searching service used in Tripal
- JBrowse: genome browser
- Nginx proxy: page to download the data files
- Blast (optional): BLAST interface to query the data
- Galaxy: data loading orchestrator for administrators
A GGA environment is deployed for each different species at https://hostname/sp/genus_species/. Multiple strains can have the same species and are deployed in the same GGA environment.
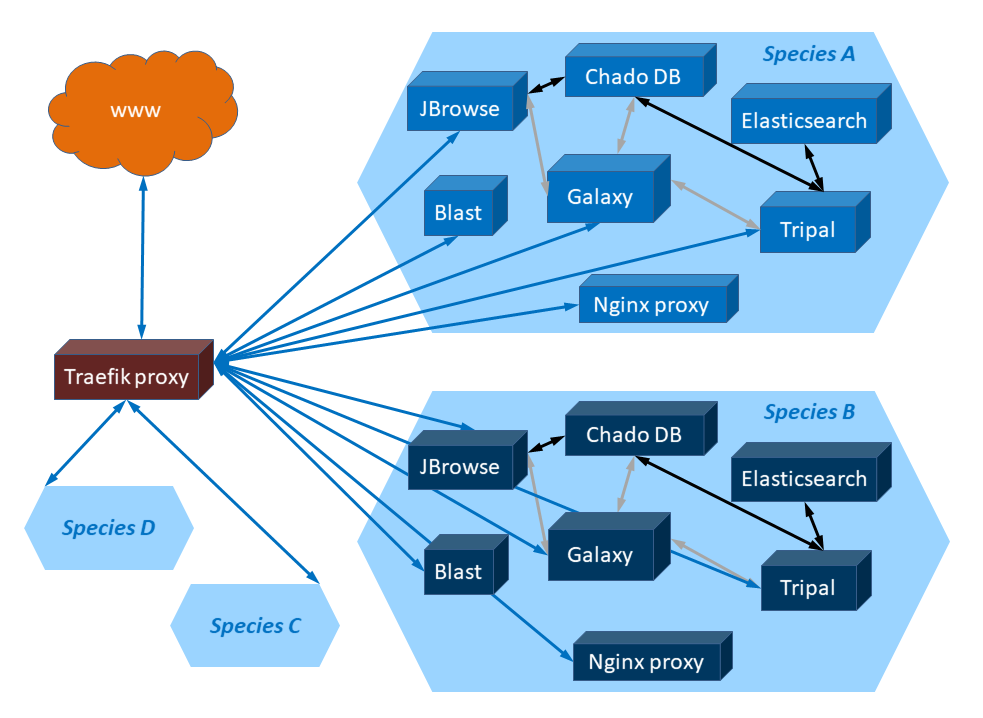 Figure : Schematic representation of a set of Docker containers deployed with
GGA for typical genomes. Cuboids represent Docker containers. Hexagons represent different
sets of Docker containers. Blue arrows represent HTTP traffic. Gray arrows represent
data exchange performed using Galaxy tools. Black arrows represent data exchange
inherent in applications.
Figure : Schematic representation of a set of Docker containers deployed with
GGA for typical genomes. Cuboids represent Docker containers. Hexagons represent different
sets of Docker containers. Blue arrows represent HTTP traffic. Gray arrows represent
data exchange performed using Galaxy tools. Black arrows represent data exchange
inherent in applications.
Requirements
To run the gga_load_data tools, Python 3.6 and the packages listed in requirements.txt are required.
To deploy the GGA Docker services, one or multiple hosts machines are required with Docker installed, and a swarm (for cluster management and orchestration).